Decoding the Complex World of Somatic Mutations: A Pioneering Whole-exome Sequencing analysis Benchmark Study
In the quest to understand and treat cancer more effectively, our genomics team has embarked on a groundbreaking journey. By collaborating with Dr. Shu-Jui Hsu’s team at National Taiwan University College of Medicine, we’ve delved into the intricate world of somatic mutations, those genetic alterations that occur in cancer cells but not in healthy cells. We leveraged the comprehensive tumor Whole Exome Sequencing (WES) data from the Sequencing and Quality Control Phase 2 (SEQC2) project to make a significant leap in evaluating the accuracy and robustness of somatic mutation analysis in clinical settings.
BY Jia-Hsin Huang
Our study aims to improve the detection of genetic changes in cancer cells using whole exome sequencing techniques. Recently, the FDA’s SEQC2 consortium has made available a new dataset for evaluating how well various methods detect cancer mutations from tumor DNA. This dataset is expected to improve the quality of cancer genetic data and set a new standard for research in this area. Finding reliable analysis tools is challenging, but vital for accurate results. We investigated 18 combinations of several reputable mutation caller and aligner tools for detecting specific types of changes, including single nucleotide variants (SNVs) and small insertions/deletions (INDELs) detection. This approach could significantly enhance cancer diagnosis and treatment planning. The preprint of this study is now available on BioRxiv.
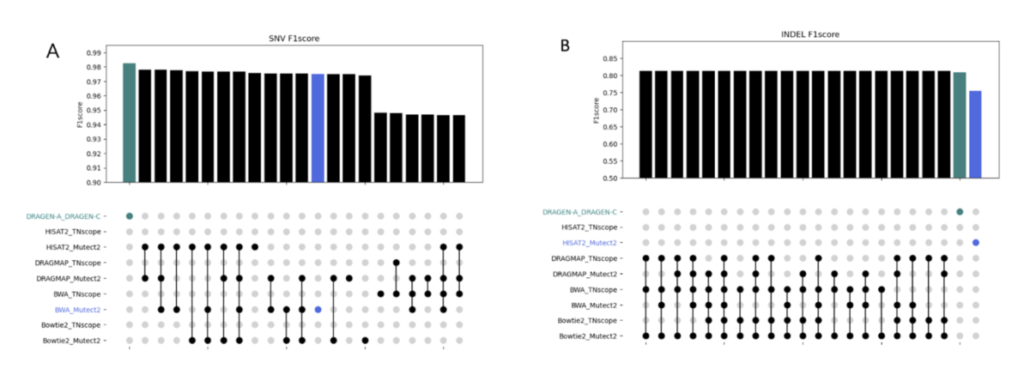
In this study, we discovered the pivotal role of mutation callers over aligners in influencing overall sensitivity in mutation detection. No single combination stood out across all tests, suggesting that combining different tools could provide the most accurate detection of genetic mutations in cancer. However, DeepVariant might not be ideal for somatic mutation analysis due to the poor performance in our tests. And the combination of BWA and Mutect2 demonstrated the best performance in open-source software categories for SNV detection. Our study highlights the benefits of using a mix of genetic analysis tools, showing improved detection of cancer mutations over using BWA_Mutect2 alone. Merging Mutect2 with various aligners slightly boosted the accuracy for single changes (SNVs), while for insertions/deletions (INDELs), these combinations outperformed DRAGEN in precision and overall accuracy.
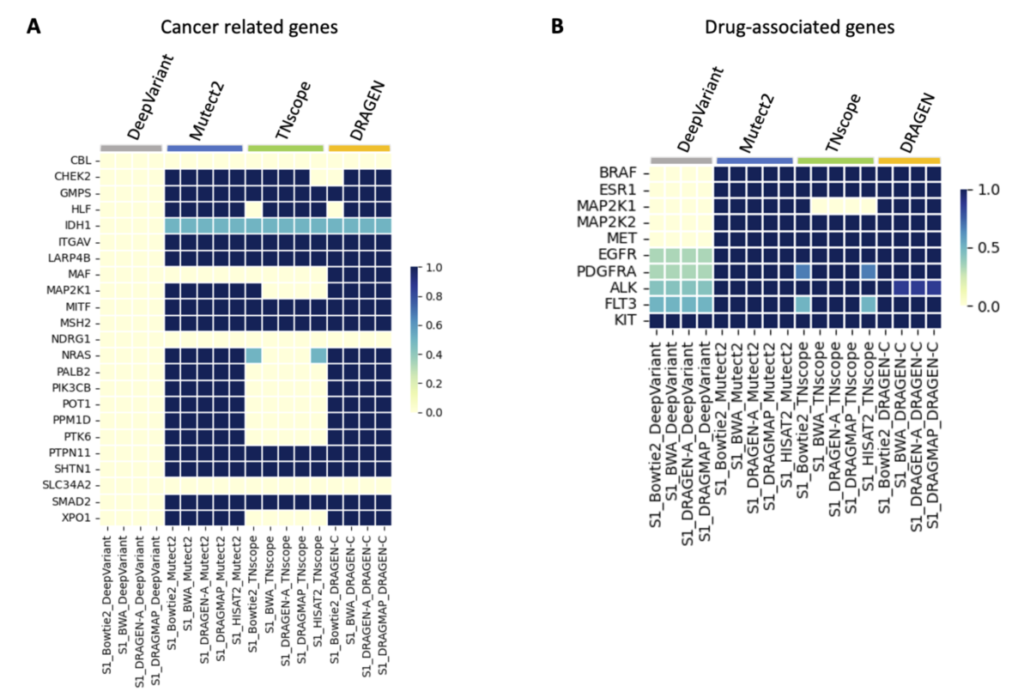
Understanding somatic mutations is not just a scientific pursuit; it has real-world implications in clinical oncology. We annotated the detected variants using the Catalogue Of Somatic Mutations In Cancer (COSMIC) to evaluate the accuracy of various variant caller tools that detect mutations in genes related to cancer. DeepVariant, in particular, struggled to identify key gene variants, suggesting it might not be the best fit for pinpointing somatic mutations crucial for cancer treatment decisions. Additionally, we found that TNscope, while generally effective, had a higher tendency to miss certain mutations. This highlights the importance of choosing the right tools for genetic analysis, as these choices can significantly influence the direction and effectiveness of a patient’s treatment.
The ability to pinpoint specific mutations aids in targeted therapies and can flag potential drug resistance, underscoring the importance of our study. We also assessed how different genetic analysis tools detect mutations linked to drug resistance and treatment choices. Key findings show that while some tools accurately identify critical mutations, others, like DeepVariant, missed more than half, which could have major implications for treatment efficacy. For instance, certain mutations that contribute to chemotherapy resistance or affect targeted therapy response were not consistently detected by all tools. This variability underscores the necessity for precise genetic testing to inform personalized medicine approaches.
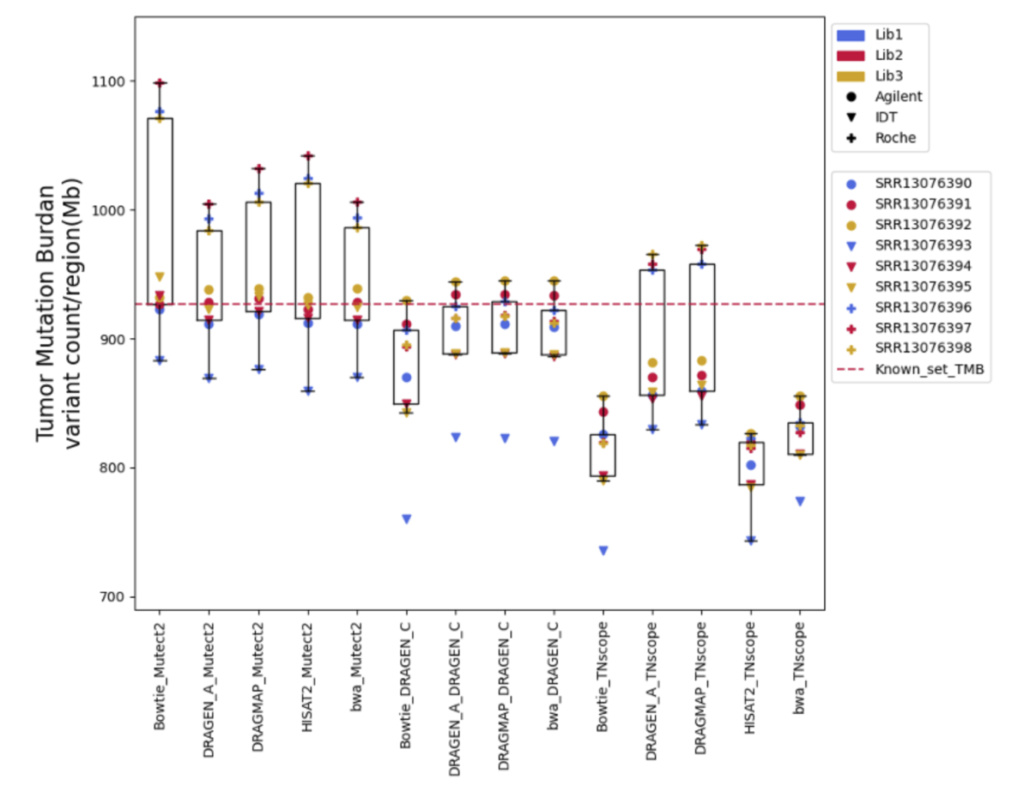
Furthermore, Tumor Mutational Burden (TMB) estimation can be indicative of a tumor’s instability and has been used as a biomarker to predict the efficacy of immunotherapy. In our research on measuring TMB, we found that different tools and sample preparation methods can produce varying results. For instance, Mutect2 often predicted higher TMB levels than what was actually present, especially in samples prepared with a specific Roche kit. Meanwhile, DRAGEN_C provided results that closely matched the expected TMB, and TNscope generally gave lower TMB estimates. These findings highlight that choosing the right tools and methods is crucial for accurate TMB measurement, which is key to making the best clinical decisions for cancer treatment.
In summary, our research offers a beacon of guidance for cancer genomic researchers, providing a detailed comparison of diverse tool combinations for tumor mutation identification. It’s a step forward in our collective mission to demystify cancer genetics and enhance the precision of medical treatments.
Reference
Somatic mutation detection workflow validity distinctly influences clinical decision
Pei-Miao Chien, Chinyi Cheng, Tzu-Hang Yuan, Yu-Bin Wang, Pei-Lung Chen, Chien-Yu Chen, Jia-Hsin Huang, Jacob Shujui Hsu. bioRxiv 2023.10.26.562640
